


OPIS PRZYPADKU
KLINICZNE I GENETYCZNE ASPEKTY ATAKSJI RDZENIOWO-MÓŻDŻKOWEJ TYPU 29
1
Student Scientific Association of Developmental Neurology, Student Scientific Society, Poznan University of Medical Sciences, Poland
2
Department of Developmental Neurology, Poznan University of Medical Sciences, Poland
Data nadesłania: 16-12-2025
Data ostatniej rewizji: 07-01-2026
Data akceptacji: 07-01-2026
Data publikacji online: 22-05-2026
Data publikacji: 02-07-2026
Autor do korespondencji
Patrycja Krysiak
Student Scientific Association of Developmental Neurology, Student Scientific Society, Poznan University of Medical Sciences, Poland
Student Scientific Association of Developmental Neurology, Student Scientific Society, Poznan University of Medical Sciences, Poland
Issue Rehabil. Orthop. Neurophysiol. Sport Promot. 2025;53(4):47-54

SŁOWA KLUCZOWE
DZIEDZINY
STRESZCZENIE
Wprowadzenie:
Ataksje rdzeniowo-móżdżkowe (SCA) to zróżnicowana grupa dziedzicznych chorób neurodegeneracyjnych, które najczęściej ujawniają się w wieku dorosłym. Jednakże, niektóre z nich, w tym SCA29, mają swój początek już w dzieciństwie. Choroba ta jest spowodowana przez dominujące warianty patogenne w genie ITPR1, który koduje kanał wapniowy o wysokiej ekspresji w komórkach Purkinjego. Klinicznie SCA29 charakteryzuje się wczesną ataksją móżdżkową, globalnym opóźnieniem ruchowym i hipotonią mięśniową w okresie niemowlęcym.
Cel:
Celem niniejszego opisu przypadku jest przedstawienie szczegółowego opisu klinicznego oraz postępowania rehabilitacyjnego u pacjenta z SCA29.
Materiał i metody:
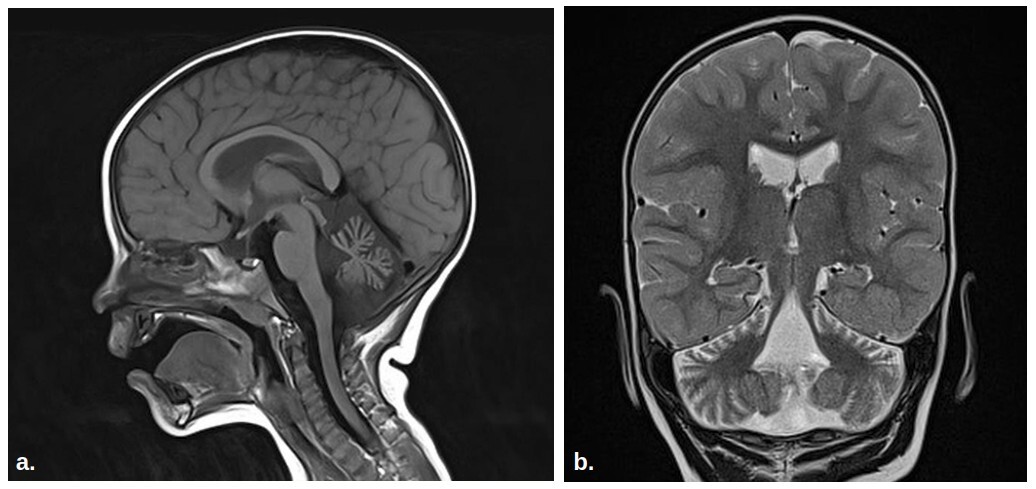
Pacjent to 5-letni chłopiec, urodzony o czasie z ciąży przebiegającej bez powikłań. Od okresu niemowlęcego występowała u niego hipotonia, opóźnione osiąganie kolejnych etapów rozwoju oraz ograniczona reakcja na bodźce wzrokowe. Od 4. roku życia chodzi wyłącznie z obustronną asekuracją. Rozwój funkcji poznawczych pacjenta jest prawidłowy, mowa dyzartryczna. Rezonans magnetyczny wykazał atrofię móżdżku, co jest istotną cechą SCA29. Sekwencjonowanie całego eksomu metodą trio zidentyfikowało patogenny heterozygotyczny wariant zmiany sensu w genie ITPR1: c.722G>A p.(Arg241Lys). Wariant ten nie występował u żadnego z rodziców, co potwierdza jego charakter de novo. Wynik badania genetycznego w korelacji z badaniami klinicznymi pozwolił na postawienie diagnozy SCA29.
Wnioski:
SCA29 jest ultrarzadkim zaburzeniem, którego objawy kliniczne pokrywają się z objawami innych schorzeń neurologicznych, utrudniając przeprowadzenie diagnostyki różnicowej. U każdego dziecka wykazującego objawy ataksji o nieznanej etiologii należy rozważyć przeprowadzenie badań genetycznych. Wcześnie postawiona diagnoza ma szczególne znaczenie, ponieważ szybko wdrożona interdyscyplinarna rehabilitacja może poprawić rozwój dziecka i umożliwić uzyskanie dostępu do grup wsparcia dla pacjentów.
Ataksje rdzeniowo-móżdżkowe (SCA) to zróżnicowana grupa dziedzicznych chorób neurodegeneracyjnych, które najczęściej ujawniają się w wieku dorosłym. Jednakże, niektóre z nich, w tym SCA29, mają swój początek już w dzieciństwie. Choroba ta jest spowodowana przez dominujące warianty patogenne w genie ITPR1, który koduje kanał wapniowy o wysokiej ekspresji w komórkach Purkinjego. Klinicznie SCA29 charakteryzuje się wczesną ataksją móżdżkową, globalnym opóźnieniem ruchowym i hipotonią mięśniową w okresie niemowlęcym.
Cel:
Celem niniejszego opisu przypadku jest przedstawienie szczegółowego opisu klinicznego oraz postępowania rehabilitacyjnego u pacjenta z SCA29.
Materiał i metody:
Pacjent to 5-letni chłopiec, urodzony o czasie z ciąży przebiegającej bez powikłań. Od okresu niemowlęcego występowała u niego hipotonia, opóźnione osiąganie kolejnych etapów rozwoju oraz ograniczona reakcja na bodźce wzrokowe. Od 4. roku życia chodzi wyłącznie z obustronną asekuracją. Rozwój funkcji poznawczych pacjenta jest prawidłowy, mowa dyzartryczna. Rezonans magnetyczny wykazał atrofię móżdżku, co jest istotną cechą SCA29. Sekwencjonowanie całego eksomu metodą trio zidentyfikowało patogenny heterozygotyczny wariant zmiany sensu w genie ITPR1: c.722G>A p.(Arg241Lys). Wariant ten nie występował u żadnego z rodziców, co potwierdza jego charakter de novo. Wynik badania genetycznego w korelacji z badaniami klinicznymi pozwolił na postawienie diagnozy SCA29.
Wnioski:
SCA29 jest ultrarzadkim zaburzeniem, którego objawy kliniczne pokrywają się z objawami innych schorzeń neurologicznych, utrudniając przeprowadzenie diagnostyki różnicowej. U każdego dziecka wykazującego objawy ataksji o nieznanej etiologii należy rozważyć przeprowadzenie badań genetycznych. Wcześnie postawiona diagnoza ma szczególne znaczenie, ponieważ szybko wdrożona interdyscyplinarna rehabilitacja może poprawić rozwój dziecka i umożliwić uzyskanie dostępu do grup wsparcia dla pacjentów.
REFERENCJE (20)
1.
Coarelli G, Coutelier M, Durr A. Autosomal dominant cerebellar ataxias: new genes and progress towards treatments. Lancet Neurol 2023; 22: 735–749.
2.
Soong BW, Morrison PJ. Spinocerebellar ataxias. Handb Clin Neurol 2018; 155: 143–174.
3.
Coarelli G, Wirth T, Tranchant C, Koenig M, Durr A, Anheim M. The inherited cerebellar ataxias: an update. J Neurol 2023; 270: 208–222.
4.
Sullivan R, Yau WY, O’Connor E, Houlden H. Spinocerebellar ataxia: an update. J Neurol 2018; 266: 533–544.
5.
Chien HF, Zonta MB, Chen J, et al. Rehabilitation in patients with cerebellar ataxias. Arq Neuropsiquiatr 2022; 80: 306–315.
6.
Brooker SM, Edamakanti CR, Akasha SM, Kuo S, Opal P. Spinocerebellar ataxia clinical trials: opportunities and challenges. Ann Clin Transl Neurol 2021; 8: 1543–1556.
7.
Matsugi A, Bando K, Kondo Y, et al. Effects of physiotherapy on degenerative cerebellar ataxia: a systematic review and meta-analysis. Front Neurol 2025; 15: 1491142. DOI: 10.3389/fneur.2024.1491142.
8.
Paulson HL, Shakkottai VG, Clark HB, Orr HT. Polyglutamine spinocerebellar ataxias – from genes to potential treatments. Nat Rev Neurosci 2017; 18: 613–626.
10.
Huang L, Warman-Chardon J, Carter MT, et al. Correction to: missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia. Orphanet J Rare Dis 2022; 17: 143. DOI: 10.1186/s13023-022-02297-7.
11.
Romaniello R, Pasca L, Panzeri E, et al. Superior cerebellar atrophy: an imaging clue to diagnose ITPR1-related disorders. Int J Mol Sci 2022; 23: 6723. DOI: 10.3390/ijms23126723.
12.
Ando H, Hirose M, Mikoshiba K. Aberrant IP3 receptor activities revealed by comprehensive analysis of pathological mutations causing spinocerebellar ataxia 29. Proc Natl Acad Sci U S A 2018; 115: 12259–12264.
13.
Zambonin JL, Bellomo A, Ben-Pazi H, et al. Spinocerebellar ataxia type 29 due to mutations in ITPR1: a case series and review of this emerging congenital ataxia. Orphanet J Rare Dis 2017; 12: 121. DOI: 10.1186/s13023-017-0672-7.
14.
Casey JP, Hirouchi T, Hisatsune C, et al. A novel gain-of-function mutation in the ITPR1 suppressor domain causes spinocerebellar ataxia with altered Ca2+ signal patterns. J Neurol 2017; 264: 1444–1453.
15.
Shimobayashi E, Kapfhammer JP. Calcium signaling, PKC Gamma, IP3R1 and CAR8 link spinocerebellar ataxias and Purkinje cell dendritic development. Curr Neuropharmacol 2018; 16: 151–159.
16.
Huang L, Warman-Chardon J, Carter MT, et al. Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia. Orphanet J Rare Dis 2012; 7: 67. DOI: 10.1186/1750-1172-7-67.
17.
Ciaccio C, Taddei M, Pantaleoni C, et al. Phenotypic spectrum and natural history of gillespie syndrome. An updated literature review with 2 new cases. Cerebellum 2024; 23: 2655–2670.
18.
Tipton PW, Guthrie K, Strongosky A, Reimer R, Wszolek ZK. Spinocerebellar ataxia 15: a phenotypic review and expansion. Neurol Neurochir Pol 2017; 51: 86–91.
19.
Radmard S, Zesiewicz TA, Kuo SH. Evaluation of cerebellar ataxic patients. Neurol Clin 2023; 41: 21–44.
20.
Lanza G, Casabona JA, Bellomo M, et al. Update on intensive motor training in spinocerebellar ataxia: time to move a step forward? J Int Med Res 2019; 48: 300060519854626. DOI: 10.1177/0300060519854626.
Udostępnij
| ISSN: | 2300-0767 |
Przetwarzamy dane osobowe zbierane podczas odwiedzania serwisu. Realizacja funkcji pozyskiwania informacji o użytkownikach i ich zachowaniu odbywa się poprzez dobrowolnie wprowadzone w formularzach informacje oraz zapisywanie w urządzeniach końcowych plików cookies (tzw. ciasteczka). Dane, w tym pliki cookies, wykorzystywane są w celu realizacji usług, zapewnienia wygodnego korzystania ze strony oraz w celu monitorowania ruchu zgodnie z Polityką prywatności. Dane są także zbierane i przetwarzane przez narzędzie Google Analytics (więcej).
Możesz zmienić ustawienia cookies w swojej przeglądarce. Ograniczenie stosowania plików cookies w konfiguracji przeglądarki może wpłynąć na niektóre funkcjonalności dostępne na stronie.
Możesz zmienić ustawienia cookies w swojej przeglądarce. Ograniczenie stosowania plików cookies w konfiguracji przeglądarki może wpłynąć na niektóre funkcjonalności dostępne na stronie.