


CASE STUDY
CLINICAL AND GENETIC INSIGHTS INTO SPINOCEREBELLAR ATAXIA TYPE 29
1
Student Scientific Association of Developmental Neurology, Student Scientific Society, Poznan University of Medical Sciences, Poland
2
Department of Developmental Neurology, Poznan University of Medical Sciences, Poland
Submission date: 2025-12-16
Final revision date: 2026-01-07
Acceptance date: 2026-01-07
Online publication date: 2026-05-22
Publication date: 2026-07-02
Corresponding author
Patrycja Krysiak
Student Scientific Association of Developmental Neurology, Student Scientific Society, Poznan University of Medical Sciences, Poland
Student Scientific Association of Developmental Neurology, Student Scientific Society, Poznan University of Medical Sciences, Poland
Issue Rehabil. Orthop. Neurophysiol. Sport Promot. 2025;53(4):47-54

KEYWORDS
TOPICS
ABSTRACT
Introduction:
Spinocerebellar ataxias (SCAs) denote a heterogeneous group of hereditary neurodegenerative disorders, most commonly presenting in adulthood. Nevertheless, few entities have their onset in childhood, including SCA type 29 (SCA29). It is associated with dominant pathogenic variants in the ITPR1 gene, which encodes a calcium channel essential for Purkinje cell function. Clinically, SCA29 is characterized by early-onset cerebellar ataxia, global motor delay, and infantile muscular hypotonia.
Aim:
The aim of this case report is to present detailed clinical and therapeutic management of an individual affected with SCA29.
Material and methods:
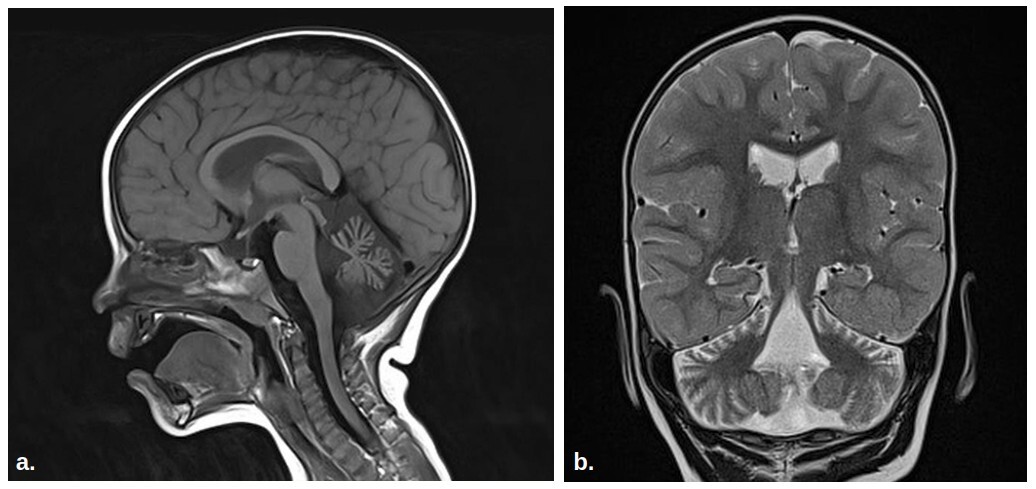
The patient is a 5-year-old boy born at term after an uneventful pregnancy. From infancy, he exhibited hypotonia, delayed attainment of milestones, and reduced responsiveness to visual stimuli. Since the age of 4 years, he has been walking only with bilateral support. The patient’s cognitive development is normal, speech is inadequate for the age, dysarthric. Magnetic resonance imaging revealed cerebellar hypoplasia, a significant feature of SCA29. Trio-based whole exome sequencing identified a pathogenic heterozygous missense variant in the ITPR1 gene: c.722G>A p.(Arg241Lys). The variant was absent from both parents, confirming its de novo character. Genetic testing combined with clinical findings led to a diagnosis of SCA29.
Conclusions:
SCA29 is an ultra-rare disorder, which clinical manifestation overlaps with various other neurological entities, posing difficulties in differential diagnosis. Genetic analysis should be considered in any child exhibiting signs of ataxia of unknown etiology. Early diagnosis is of particular importance, as timely implemented interprofessional rehabilitation may enhance the children’s development and facilitate access to patient support groups.
Spinocerebellar ataxias (SCAs) denote a heterogeneous group of hereditary neurodegenerative disorders, most commonly presenting in adulthood. Nevertheless, few entities have their onset in childhood, including SCA type 29 (SCA29). It is associated with dominant pathogenic variants in the ITPR1 gene, which encodes a calcium channel essential for Purkinje cell function. Clinically, SCA29 is characterized by early-onset cerebellar ataxia, global motor delay, and infantile muscular hypotonia.
Aim:
The aim of this case report is to present detailed clinical and therapeutic management of an individual affected with SCA29.
Material and methods:
The patient is a 5-year-old boy born at term after an uneventful pregnancy. From infancy, he exhibited hypotonia, delayed attainment of milestones, and reduced responsiveness to visual stimuli. Since the age of 4 years, he has been walking only with bilateral support. The patient’s cognitive development is normal, speech is inadequate for the age, dysarthric. Magnetic resonance imaging revealed cerebellar hypoplasia, a significant feature of SCA29. Trio-based whole exome sequencing identified a pathogenic heterozygous missense variant in the ITPR1 gene: c.722G>A p.(Arg241Lys). The variant was absent from both parents, confirming its de novo character. Genetic testing combined with clinical findings led to a diagnosis of SCA29.
Conclusions:
SCA29 is an ultra-rare disorder, which clinical manifestation overlaps with various other neurological entities, posing difficulties in differential diagnosis. Genetic analysis should be considered in any child exhibiting signs of ataxia of unknown etiology. Early diagnosis is of particular importance, as timely implemented interprofessional rehabilitation may enhance the children’s development and facilitate access to patient support groups.
REFERENCES (20)
1.
Coarelli G, Coutelier M, Durr A. Autosomal dominant cerebellar ataxias: new genes and progress towards treatments. Lancet Neurol 2023; 22: 735–749.
2.
Soong BW, Morrison PJ. Spinocerebellar ataxias. Handb Clin Neurol 2018; 155: 143–174.
3.
Coarelli G, Wirth T, Tranchant C, Koenig M, Durr A, Anheim M. The inherited cerebellar ataxias: an update. J Neurol 2023; 270: 208–222.
4.
Sullivan R, Yau WY, O’Connor E, Houlden H. Spinocerebellar ataxia: an update. J Neurol 2018; 266: 533–544.
5.
Chien HF, Zonta MB, Chen J, et al. Rehabilitation in patients with cerebellar ataxias. Arq Neuropsiquiatr 2022; 80: 306–315.
6.
Brooker SM, Edamakanti CR, Akasha SM, Kuo S, Opal P. Spinocerebellar ataxia clinical trials: opportunities and challenges. Ann Clin Transl Neurol 2021; 8: 1543–1556.
7.
Matsugi A, Bando K, Kondo Y, et al. Effects of physiotherapy on degenerative cerebellar ataxia: a systematic review and meta-analysis. Front Neurol 2025; 15: 1491142. DOI: 10.3389/fneur.2024.1491142.
8.
Paulson HL, Shakkottai VG, Clark HB, Orr HT. Polyglutamine spinocerebellar ataxias – from genes to potential treatments. Nat Rev Neurosci 2017; 18: 613–626.
10.
Huang L, Warman-Chardon J, Carter MT, et al. Correction to: missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia. Orphanet J Rare Dis 2022; 17: 143. DOI: 10.1186/s13023-022-02297-7.
11.
Romaniello R, Pasca L, Panzeri E, et al. Superior cerebellar atrophy: an imaging clue to diagnose ITPR1-related disorders. Int J Mol Sci 2022; 23: 6723. DOI: 10.3390/ijms23126723.
12.
Ando H, Hirose M, Mikoshiba K. Aberrant IP3 receptor activities revealed by comprehensive analysis of pathological mutations causing spinocerebellar ataxia 29. Proc Natl Acad Sci U S A 2018; 115: 12259–12264.
13.
Zambonin JL, Bellomo A, Ben-Pazi H, et al. Spinocerebellar ataxia type 29 due to mutations in ITPR1: a case series and review of this emerging congenital ataxia. Orphanet J Rare Dis 2017; 12: 121. DOI: 10.1186/s13023-017-0672-7.
14.
Casey JP, Hirouchi T, Hisatsune C, et al. A novel gain-of-function mutation in the ITPR1 suppressor domain causes spinocerebellar ataxia with altered Ca2+ signal patterns. J Neurol 2017; 264: 1444–1453.
15.
Shimobayashi E, Kapfhammer JP. Calcium signaling, PKC Gamma, IP3R1 and CAR8 link spinocerebellar ataxias and Purkinje cell dendritic development. Curr Neuropharmacol 2018; 16: 151–159.
16.
Huang L, Warman-Chardon J, Carter MT, et al. Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia. Orphanet J Rare Dis 2012; 7: 67. DOI: 10.1186/1750-1172-7-67.
17.
Ciaccio C, Taddei M, Pantaleoni C, et al. Phenotypic spectrum and natural history of gillespie syndrome. An updated literature review with 2 new cases. Cerebellum 2024; 23: 2655–2670.
18.
Tipton PW, Guthrie K, Strongosky A, Reimer R, Wszolek ZK. Spinocerebellar ataxia 15: a phenotypic review and expansion. Neurol Neurochir Pol 2017; 51: 86–91.
19.
Radmard S, Zesiewicz TA, Kuo SH. Evaluation of cerebellar ataxic patients. Neurol Clin 2023; 41: 21–44.
20.
Lanza G, Casabona JA, Bellomo M, et al. Update on intensive motor training in spinocerebellar ataxia: time to move a step forward? J Int Med Res 2019; 48: 300060519854626. DOI: 10.1177/0300060519854626.
| ISSN: | 2300-0767 |
We process personal data collected when visiting the website. The function of obtaining information about users and their behavior is carried out by voluntarily entered information in forms and saving cookies in end devices. Data, including cookies, are used to provide services, improve the user experience and to analyze the traffic in accordance with the Privacy policy. Data are also collected and processed by Google Analytics tool (more).
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.